In the latest version of the HMMER website we have focused on enhancing the recognition and display of domains and motifs found in query sequences. To achieve this we added two new features to the site, additional HMM databases and simple motif detection.
Additional protein family HMM databases
We have added three additional protein family databases, in addition to the Pfam HMM library, which can be searched using hmmscan. Thus, it is now possible to search against Gene3D, Pfam, Superfamily and/or TIGRFAMs. These databases have been added due to their popularity and as the models have been produced using HMMER 3.0. Why might you want to search your sequence against different protein family databases? Well, although we are closely allied to Pfam, other databases offer a different view of the protein family world. Gene3D and Superfamily are based on the structural classifications CATH and SCOP respectively. Consequently, a match to one of these domains means that there will be a known structural homolog for that region. TIGRFAMs models are more specific compared to Pfam and are primarily designed to be used for sequence annotation. Below, is a graphical representation of the matches of the four different HMM databases, when queried with the same sequence (UniProt accession B0A027).
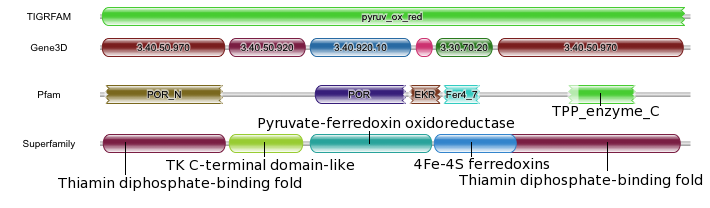
TIGRFAM has matched nearly the full length of the sequence, with the pyruv_ox_red model. If you follow the links through to the TIGRFAM site, the assigned functional annotation would be ‘pyruvate:ferredoxin (flavodoxin) oxidoreductase’ – the sequence is annotated as such in UniProt. Such oxidoreductases are required for the transfer of electrons from pyruvate to flavodoxin. Consequently, the sequences have several different functional domains to perform the task. If the annotations from Pfam, Gene3D and Superfamily are compared, there is general agreement between the domain annotations, but there are some differences. All three databases annotate the N-terminal domain. The second domains annotated by Gene3D and Superfamily are very similar to each other. However, Pfam does not currently annotate an equivalent domain. The middle two domains in Pfam (POR colored purple and EKR colored brown) and Gene3D (3.40.920.10 colored blue, 4.10.780.10 colored pink) are again very similar. However, in Superfamily, the corresponding region is represented by a single entry. The next and penultimate domain, the iron-sulfur binding domain, is detected by all three databases, but is longest in Superfamily and shortest in Pfam. The final, C-terminal domains from Superfamily and Gene3D are, again very similar. Both databases indicate that the very N-terminal and C-terminal domains arise from the same structural superfamily. The equivalent domain from Pfam is a partial match against the model. This family, TPP_enzyme_C is in the same clan as POR_N, again showing the relationship between the N- and C- terminal domains.
You will probably also notice that the Gene3D and Superfamily domains all have curved edges in the domain graphic, where as both TIGRFAM and Pfam have some jagged ends. The difference is that for Pfam and TIGRFAM we indicate when the match does not match the full length of the HMM – only when it matched the first position and/or the last position in the HMM will it get a curved end. However, the same approach cannot be applied to Gene3D and Superfamily as both of these domain databases employ post-processing to the results from hmmscan. This post processing is required as these databases use multiple HMMs to model a single structural superfamily and consequently can result in many overlapping matches that represent the same superfamily – note that both databases allow some limited overlap between the termini of domains, as shown above for Superfamily between the two C-terminal domains. Furthermore, due to the post processing we have altered the results table, a little for Gene3D and more substantially for Superfamily. Nevertheless, the profile HMM match data is still available under the advanced tabular results view. We have also removed the ability to adjust the cut-offs for these databases as the post-processing uses an internal threshold that would negate any user threshold adjustments.
other sequence motifs
The other area of sequence motif detection that we have added is the detection of motifs other than protein families or domains, namely disorder, transmembrane, signal peptide and coiled-coils. When a single sequence is submitted to the site (using hmmscan, phmmer or jackhmmer), we search it for these motifs using three pieces of software –IUPred for disorder prediction, ncoils for coiled-coil prediction and Phobius for signal peptide and transmembrane predictions. Only when motifs are predicted by one of the methods are the data graphically represented. The results are dynamically loaded into the page as soon as they have finished, so the ordering below the domain graphic can change, depending on which method finished first.
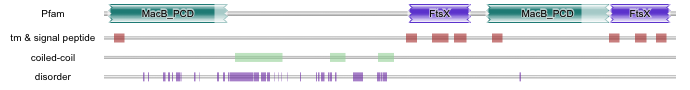
In the example shown above (UniProt accession F0EMD7), the region not covered by Pfam domains contains a lot of disordered sequence (which also overlaps with coiled-coil predictions). This region of sequence is very glutamine rich, which is why this region has probably not been represented by a Pfam entry. However, the presence of disorder does not mean that the region is not functional and/or unconserved. There are Pfam entries that cover large stretches of predicted disordered sequence, such as Hox9 activation region (Pfam accession PF04617). Below is a graphical representation of the Human Hox A9 (UniProt accession P31269), clearly showing the overlap between the disorder prediction and the Pfam domain.

These “non-domain” motif annotations are currently not available via the API, but we will be adding them to the API repertoire and putting the appropriate documentation in place shortly. We hope these additional sequence annotations will help you further understand your results!