It has been a couple of weeks now since we released jackhmmer on the HMMER website and so far (touch wood etc…), it seems to be performing as we had hoped – here on ‘the farm’ we are getting very excited with the results we are observing.
what is jackhmmer?
Jackhmmer is the iterative search method in the HMMER package, analogous to PSI-BLAST. A typical jackhmmer search starts with a single protein sequence. This sequence is searched against a target protein sequence database (such as UniProt or NR) by converting the sequence to a profile hidden Markov model (HMM), using a substitution matrix and affine gap penalties. The target sequences that score above the inclusion threshold from this first search are then aligned and used to construct a second profile HMM. In this and subsequent iterations, the conservation pattern specific to this alignment is used to determine the probabilities of seeing different amino acids, a deletion or an insertion at each position in the profile HMM. This second profile HMM is then searched against the same target database and will typically find more distant homologs to the original query sequence. These new results can then be aligned as before and used to initiate another search (or iteration) and so on. Sometimes, no additional sequences can be found between iterations and the search is deemed to have converged.
website jackhmmer additions
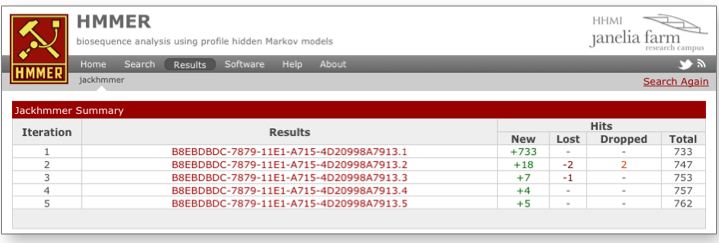
The layering of a web interface on top of jackhmmer has enabled us to provide a few additional features that do not naturally lend themselves to the command line version. The first is the ability to summarize the results (Figure 1) from each iteration to allow access to each set of results with the same repertoire of views (score, taxonomy and domain based) for their analysis. This table lists the counts of sequences that are either found, new, lost (no longer found in the results) or dropped (fall below inclusion threshold, but still found in the reported insignificant results).
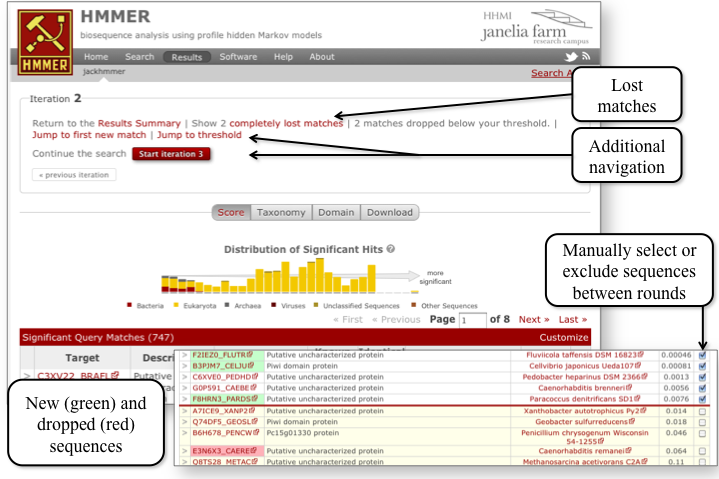
After navigating through to the results from an iteration, you can access the details of hits gained, lost or dropped. The second is the ability to manually select sequences that are to be included or excluded from the alignment used to produce the profile HMM in subsequent iterations.
so, why are we excited?
jackhmmer unleashes the power of the sophisticated probabilistic models that underpin profile HMMs. If you are trying to accurately find relationships between sequences, then profile HMMs are the most sensitive tools to use – that is why so many protein family databases use them. But how does this translate to a biologist? I have looked at several Pfam families that do not currently match a determined three-dimensional structure. In many cases, simply iterating the Pfam model using jackhmmer, through 2 or 3 iterations against UniProt and then searching with the final profile HMM against the PDB (structure) database frequently identifies significant matches to part of, or all of that family. Note, there is an option to use the results from one search to start a hmmsearch, so it really is a simple process (figure 3). Knowing the likely structure of a sequence can generate functional hypothesis and guide many different types of wet lab experiments.
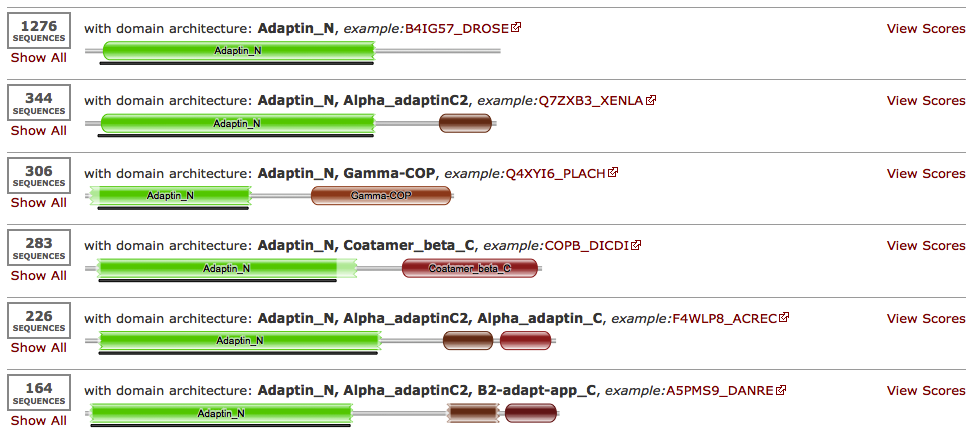
As an example, searching with the Pfam family Coatamer_beta_C against the UniProt sequence database using jackhmmer for just 2 iterations and then once with the profile HMM against PDB allows the identification of the structure 1QTS. The structure of 1QTS is the carboxyl terminal half of full length AP2A2_MOUSE sequence, the mouse AP-2 alpha subunit, and contains two globular domains, called Alpha_adatinC2 and Alpha_adaptin_C in Pfam. The N-terminal region of AP2A2_MOUSE that is absent from 1QTS corresponds to the Adaptin_N domain. The domains found on the full length sequence are found below, Figure 4. Alpha adaptins are subunits in the important AP-2 adaptor heterotetramer involved in Golgi trafficking. Specifically, this complex regulates the clathrin-bud formation at the cell surface by recruiting clathrin trimers to the plasma membrane and by selecting certain membrane proteins for inclusion within the developing clathrin-coat structure. These functions are performed by discrete subunits of the adaptor heterotetramer. The carboxyl-terminal region (termed appendage) of the AP-2 alpha subunit is thought to regulate the translocation of several endocytic accessory proteins to the bud site.
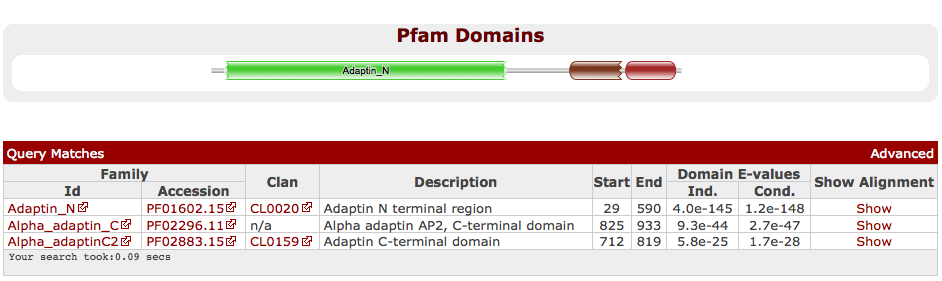
Examining the domain architecture of the sequences matched by Adaptin_N, reveals that the most abundant associated C-terminal domains are Alpha_adaptinC2/Alpha_adaptinC (the families found in 1QTS), B2-adapt-app_C, Coatamer_beta_C (the family that started this search) and Gamma-COP. There are also over 1200 sequences where the C-terminal region is not currently covered by a Pfam entry - a likely source of new families.
The RCSB PDB website provides list of similar structures that have been found using the jFATCAT-rigid algorithm. The list of similar structures to 1QTS includes other structures that are matched by the Alpha_adaptinC2/ Alpha_adaptin_C families and, interestingly, structures are matched by Gamma-COP family. This combined with the jackhmmer results and common domain architecture indicates that the Pfam annotated C-terminal regions of these Adaptin_N-containing sequences shown in figure 5 are all evolutionarily related. Furthermore, the Gamma-COP and Coatamer_beta_C domains need to split, based on the structures and would be consistent with the other domains Alpha_adaptinC2. Alpha_adaptinC and B2-adapt-app_C.
After splitting the Gamma-COP and Coatamer_beta_C Pfam families into domains, they can be grouped into Pfam clans. There should be one new Pfam clan, representing Alpha_adaptin_C-like domains. The Alpha_adaptinC2-like regions should be in the E-set clan.
summary
The set of searches described above, which started from a jackhmmer search, has allowed:
- Identification of a structural homolog for Coatamer_beta_C
- Identification of related Pfam entries (Coatamer_beta_C, Gamma-COP, Alpha_adaptinC2/ Alpha_adaptin_C)
- Likely new Pfam entries on those sequeces lacking any C-terminal domains
- Refinement of the Pfam domain boundaries for two families (which will result in two additional families)
- Proposal of a new Pfam clan
- Addition to an existing Pfam clan
Admittedly, some of this could have been determined from analyzing the structures in detail, but would not have included the relationship of Coatamer_beta_C. However, the point is that the handful of searches performed to get to these results each took a couple of seconds and allowed me to rapidly explore the sequence/structure space, without time to get distracted. It has taken me far longer to write this blog than it took to perform the analysis! On behalf of the HMMER team, I hope you find jackhmmer useful!